

Following its previous whole-genome tracing study of Brucella abortus in cattle from Zhejiang Province , the bioinformatics team at Macro & Micro-Test has once again contributed to a collaborative genomic investigation of Brucella. The study, entitled “Identification and Genome Phylogenetic Analysis of Three Brucella abortus Strains From Sheep, Yak, and Cow in Qinghai, China”, was published in Transboundary and Emerging Diseases (2026, IF = 3.4). The research was jointly conducted by the Lanzhou Veterinary Research Institute, the Qinghai Institute for Endemic Disease Prevention and Control, the National Institute for Communicable Disease Control and Prevention (China CDC), and several other institutions.
Macro & Micro-Test’s bioinformatics team is listed among the co-authors and was responsible for whole-genome comparative analysis, phylogenetic reconstruction, and data visualization.
Research Background
Qinghai Province is one of the regions most heavily affected by brucellosis in China, with the reported incidence in humans increasing from 2.45 per 100,000 in 2019 to 34.86 per 100,000 in 2023. Previous molecular epidemiological studies have focused primarily on B. melitensis, while systematic genomic investigations of B. abortus—which circulates among cattle, yaks, and wildlife—have remained limited.
In this study, three B. abortus strains were isolated from the spleens of aborted livestock fetuses collected in Qinghai Province during 2015–2016. The isolates originated from sheep (BA0611), yak (BAHYS), and cattle (BAQHM). Two previously characterized strains, QH5 and QH22, were included as lineage controls. The study fills an important knowledge gap regarding the transmission dynamics of bovine Brucella among multiple host species on the Qinghai–Tibet Plateau.
Key Findings
Confirmation of a Cross-Species Cluster Infection
The three strains isolated from sheep, yak, and cattle (BA0611, BAHYS, and BAQHM) were all identified as sequence type ST2 through multilocus sequence typing (MLST). Genomic analysis revealed that these seemingly independent infections across different host species actually originated from a common source, representing a single cluster infection event.
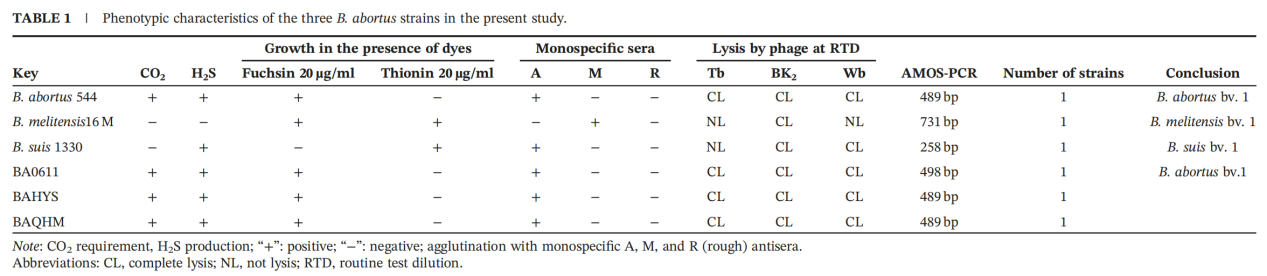
This finding challenges the conventional assumption that distinct host species harbor genetically distinct Brucella strains and highlights the substantial risk of cross-species transmission within mixed grazing systems involving cattle, sheep, and yaks in Qinghai’s pastoral regions.
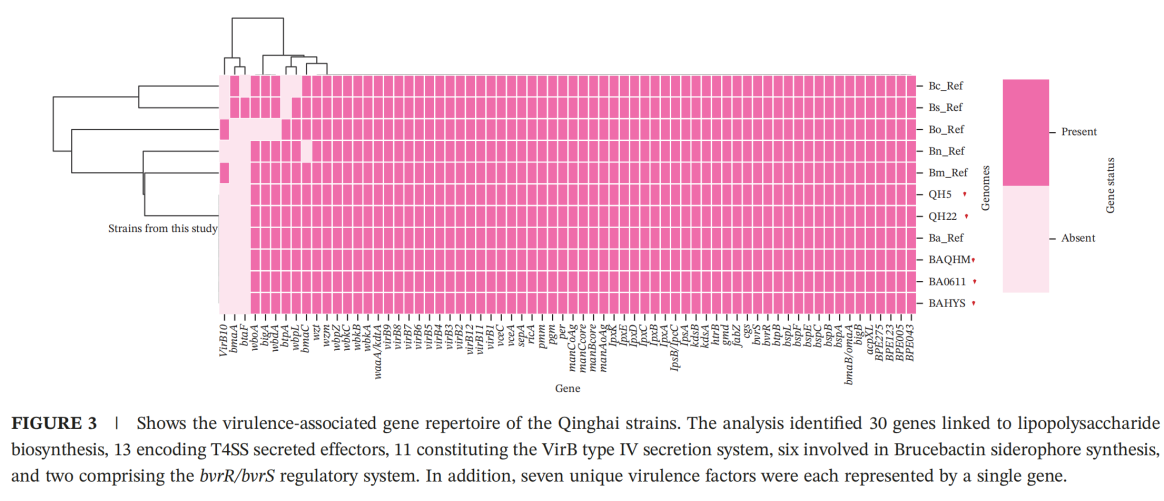
Discovery of a Unique Virulence-Deficient Lineage
Although the three strains retained the majority of known virulence-associated genes (69 in total), they consistently lacked three key genes: bmaA, btpB, and virB10. Despite these deficiencies, the strains have maintained stable circulation among animal populations on the Qinghai–Tibet Plateau.
This observation suggests either the emergence of compensatory evolutionary mechanisms in locally circulating strains or selective adaptation driven by the unique environmental conditions of the high-altitude plateau. These findings provide new insights into the pathogenicity and host adaptation strategies of Brucella
Coexistence of Multiple Lineages on the Qinghai–Tibet Plateau
Core-genome SNP (cgSNP)-based phylogenetic analysis revealed that the Qinghai strains can be divided into two distinct subclades:
Subclade I: The Qinghai animal-derived strains (BA0611, BAHYS, and BAQHM) clustered closely with a Tibetan yak strain (XZ19-1) and a strain from Russia.
Subclade II: Human-derived and marmot-derived strains from Qinghai exhibited closer genetic relationships with strains originating from northern China, including Ningxia, Heilongjiang, and Hebei.
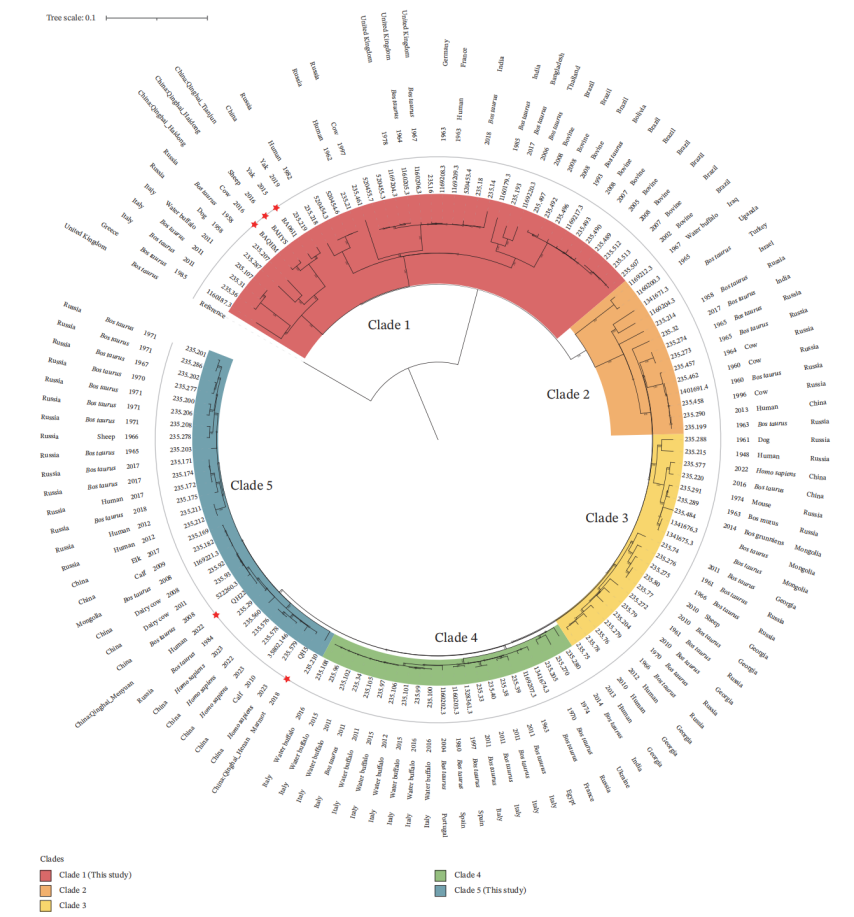
These findings indicate that Brucella transmission on the Qinghai–Tibet Plateau is not driven by a single transmission chain but rather involves at least two independent evolutionary lineages, potentially associated with historical livestock movement patterns, including interregional animal transport and cross-border trade.
Technical Contributions from Macro & Micro-Test
This study provides the first whole-genome evidence of B. abortus circulation across three major domestic livestock hosts on the Qinghai–Tibet Plateau. It expands the known host spectrum in the region and establishes a valuable genetic baseline for cross-host surveillance, source tracing, and precision control strategies under the One Health framework.
Macro & Micro-Test contributed comprehensive WGS data analysis, high-resolution cgSNP phylogenetic reconstruction, and virulence/resistance-associated functional annotation. These capabilities complement the company’s earlier Zhejiang seven-strain tracing study: while the Zhejiang investigation revealed the southward introduction of northern Brucella lineages, the present study uncovers the existence of locally adapted multi-host plateau lineages alongside evidence of interregional genetic exchange.
Together, these two studies demonstrate that cgSNP-level genomic resolution is indispensable for tracking low-diversity pathogens such as Brucella. They further highlight the importance of establishing a cross-regional and cross-host genomic surveillance network as a critical component of future brucellosis prevention and elimination efforts.
Post time: Jun-23-2026