
—— Collaborative Study by Zhejiang CDC, Macro & Micro-Test, and China CDC Published in Frontiers in Cellular and Infection Microbiology
Study Overview
In May 2026, Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) published a paper led by the Zhejiang Provincial Center for Disease Control and Prevention (Zhejiang CDC), with the bioinformatics team from Beijing Macro & Micro-Test Bio-Tech Co., Ltd. and the National Institute for Communicable Disease Control and Prevention (China CDC) as co-authors. The study is titled:
“Identification and phylogenetic analysis of seven Brucella abortus strains in Zhejiang, China.”

This study represents the first systematic, whole-genome–based phylogenetic traceability analysis of Brucella abortus (B. abortus) in Zhejiang Province, China. The team analyzed seven isolates collected from 2015 to 2025 (four human-origin and three bovine-origin strains from Jinhua, Quzhou, and Ningbo). The findings provide genomic evidence for the origin and transmission routes of this “northern dominant species” in an atypical southern epidemic region of eastern China.
Background and Significance
Brucellosis is a zoonotic disease caused by bacteria of the genus Brucella. Brucella abortus primarily infects cattle but can also cause disease in humans. In China, brucellosis shows marked geographical variation: the highest incidence occurs in northern provinces (e.g., Inner Mongolia, Shanxi, Heilongjiang). By contrast, southern provinces, including Zhejiang, have historically been dominated by Brucella melitensis, with very few reported cases of B. abortus. This regional disparity makes the genetic characterization and source tracing of B. abortus in Zhejiang a key public health priority.
Methods and Key Findings
The research team adopted a multi-pronged strategy combining molecular biology and bioinformatics:
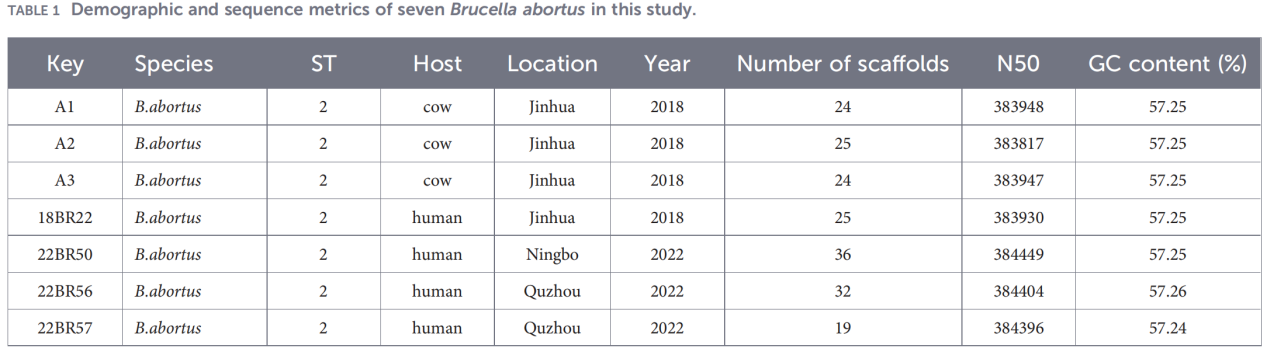
1.Pathogen identification and basic typing
BCSP-31 gene PCR and AMOS-PCR confirmed that all seven isolates were B. abortus.
Multilocus sequence typing (MLST) based on nine housekeeping genes revealed that all isolates belonged to sequence type ST2, indicating high genetic homogeneity among the circulating B. abortus strains in Zhejiang.
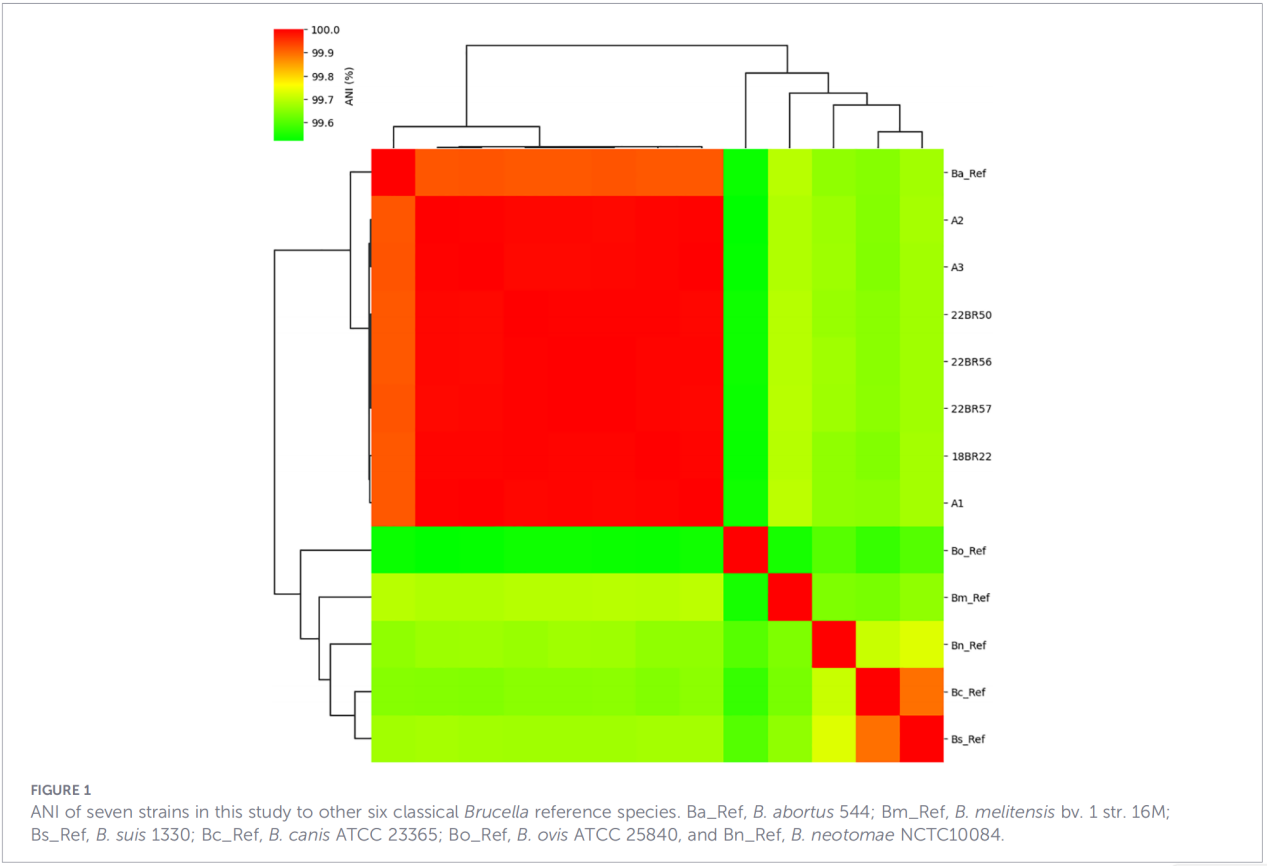
2.Whole-genome characterization
Whole-genome sequencing was performed on the Illumina NovaSeq platform. Average nucleotide identity (ANI) analysis showed that the Zhejiang isolates shared up to 99.99% similarity with the reference strain B. abortus 544.
Pan-genome analysis revealed a highly conserved population: 3,084 core genes were identified, along with only 10 shell genes, and no soft core or cloud genes were detected.
3.Virulence and antimicrobial resistance gene profiles
A total of 68 virulence-related factors were predicted, covering classical pathways such as LPS biosynthesis, the T4SS secretion system, and the BvrR-BvrS two-component regulatory system.Notably, all isolates lacked the adhesin genes bmaA and btaF. Resistance gene analysis detected only the mprF gene in the CARD database, with no other resistance determinants identified.
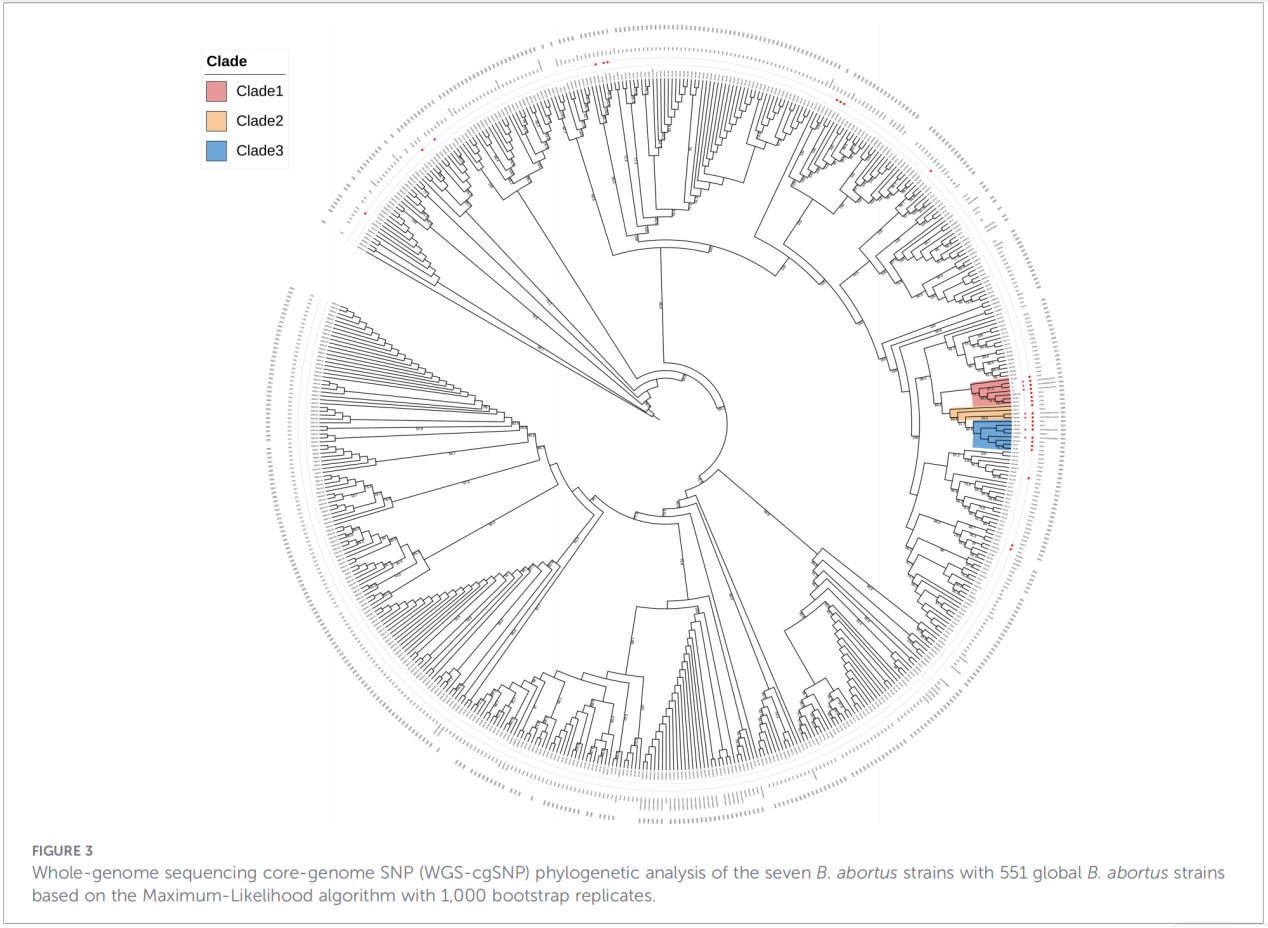
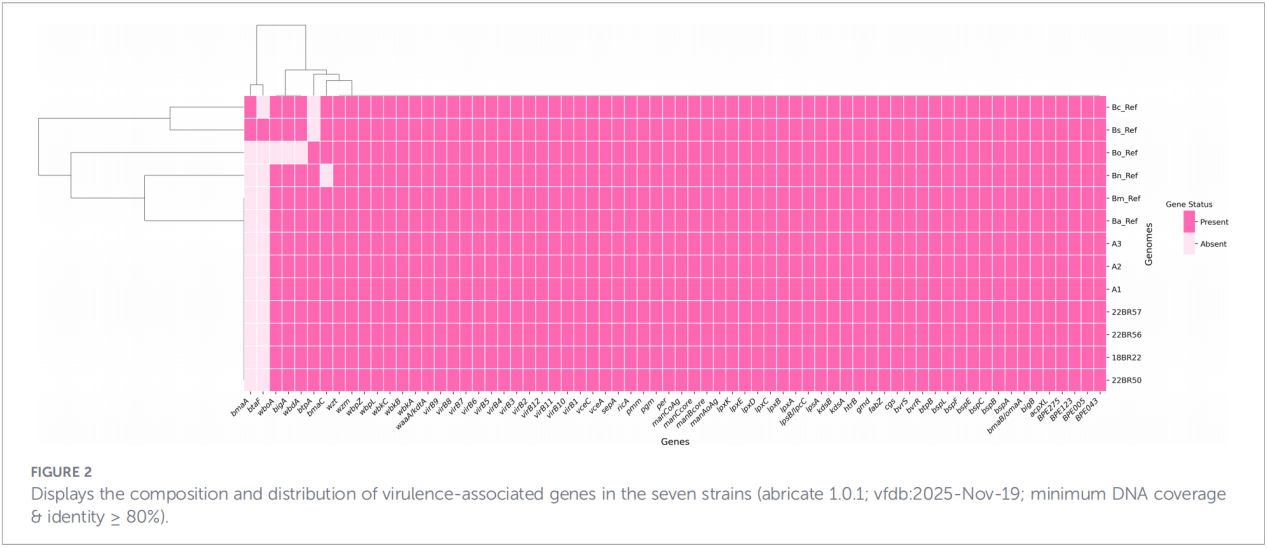 4.Phylogenetic reconstruction and transmission tracing
4.Phylogenetic reconstruction and transmission tracing
Core-genome single-nucleotide polymorphism (cgSNP) analysis placed the Zhejiang isolates at a specific position in the global phylogenetic tree. The results showed that the Zhejiang strains form a monophyletic group together with strains from Russia, Mongolia, and several northern Chinese provinces (Ningxia, Heilongjiang, Inner Mongolia, Hebei, Gansu, Beijing). This group further splits into three distinct sub clades (Clade 1–3), suggesting multiple independent introduction events.
Conclusions and Implications
This study provides the first high precision genomic dataset of B. abortus in Zhejiang Province and yields several key conclusions:
- Clear genetic background – The B. abortus strains circulating in Zhejiang belong to ST2, are genomically highly conserved, and represent a typical bovine brucellosis lineage.
2. Evidence of cross regional transmission – Phylogenetic analysis does not support the existence of an independent endemic lineage in Zhejiang. Instead, the data strongly suggest that these strains originated from northern China and may share a common evolutionary background with strains from Russia and Mongolia. The presence of three sub clades implies multiple separate introduction events.
3. Public health implications – The findings underscore the value of genomic surveillance for brucellosis even in traditionally non endemic regions such as Zhejiang. Although the current case count is low, high resolution tools like cgSNP can effectively trace the source of imported outbreaks and provide scientific evidence to interrupt transmission chains associated with inter provincial livestock transport.
This work not only fills a research gap in Zhejiang Province but also provides new baseline data for pathogen surveillance and risk assessment of brucellosis in the Yangtze River Delta region.
Paper Information:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identification and phylogenetic analysis of seven Brucella abortus strains in Zhejiang, China. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Post time: Jun-10-2026